Workflows¶
To orchestrate tools and help users with their analyses, several workflows populate ASaiM framework. They formally orchestrate tools in a defined order and with defined parameters, but they are customizable (tools, order, parameters).
Analysis of raw metagenomic or metatranscriptomic shotgun data¶
The workflow quickly produces, from raw metagenomic or metatranscriptomic shotgun data, accurate and precise taxonomic assignations, wide extended functional results and taxonomically related metabolism information
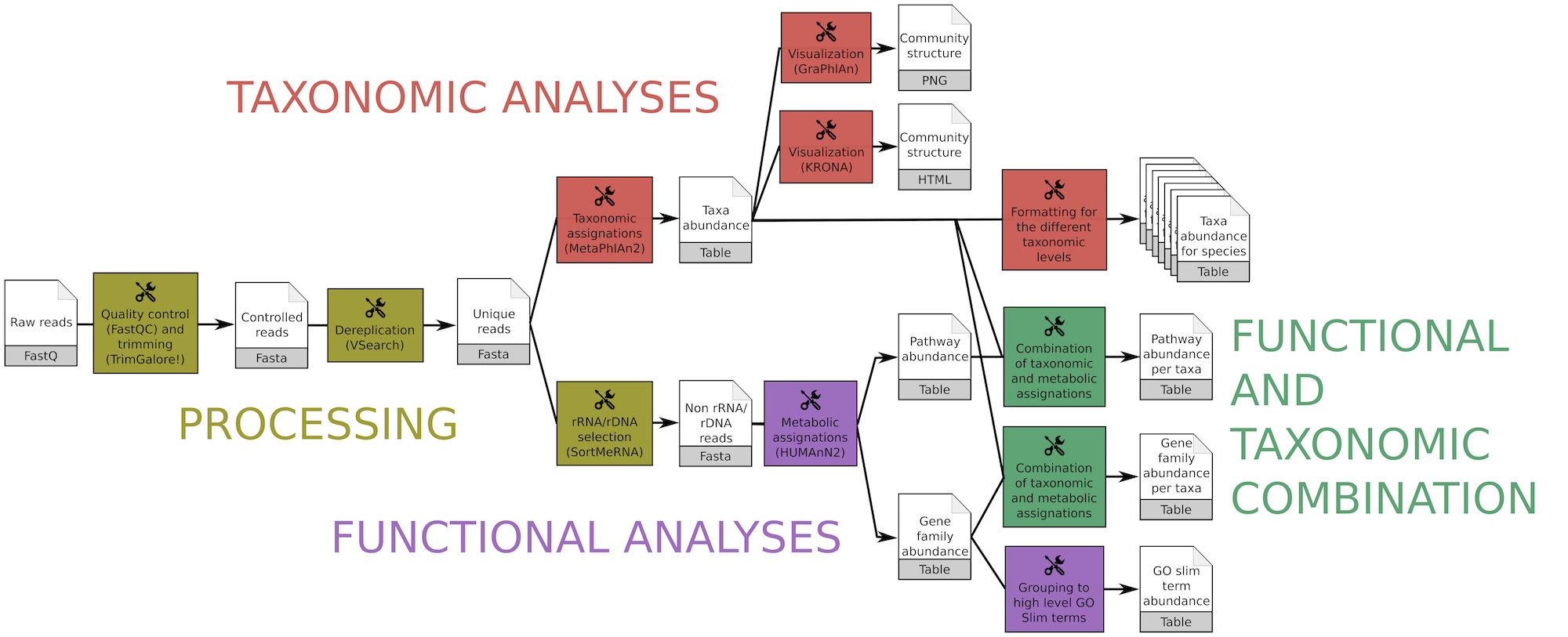
Main ASaiM workflow to analyze raw sequences. Image available under CC-BY license (https://doi.org/10.6084/m9.figshare.5371396.v3)
This workflow consists of
- Processing with quality control/trimming (FastQC and Trim Galore!) and dereplication (VSearch [13])
- Taxonomic analyses with assignation (MetaPhlAn2 [15]) and visualization (KRONA [11], GraPhlAn [2])
- Functional analyses with metabolic assignation and pathway reconstruction (HUMAnN2 [1])
- Functional and taxonomic combination with developed tools combining HUMAnN2 and MetaPhlAn2 outputs
This workflow has been tested on two mock metagenomic datasets with controlled communities (See “Validation”).
Assembly of metagenomic data¶
To reconstruct genomes or to get longer sequences for further analysis, microbiota data needs to be assembled, using the recently developed metagenomics assemblers.
To help in this task, two workflows have been developed in ASaiM, each one using one of each of the well-performing assemblers [12][16][14][5][10][17][3]
MEGAHIT [7]
It is currently the most efficent computationally assembler: it has the lowest memory and time consumption [16][3][14]. It produced some of the best assemblies (irrespective of sequencing coverage) with the fewest structural errors [10] and outperforms in recovering the genomes of closely related strains [3], but has a bias towards relatively low coverage genomes leading to a suboptimal assembly of high abundant community member genomes in very large datasets [17]
MetaSPAdes [9]
Both workflows consists of
- Processing with quality control/trimming (FastQC and Trim Galore!)
- Assembly with either MEGAHIT or MetaSPAdes
- Estimation of the assembly quality statistics with MetaQUAST [8]
- Identification of potential assembly error signature with VALET
- Determination of percentage of unmapped reads with Bowtie2 [6] combined with MultiQC [4] to aggregate the results.
Analysis of metataxonomic data¶
To analyze amplicon data, the Mothur and QIIME tool suites are available to ASaiM. We integrated the workflows described in tutorials of Mothur and QIIME websites, as example of amplicon data analyses as well as support for the training material. These workflows, as any workflows available in ASaiM, can be adapted for a specific analysis or used as subworkflows by the users.
Running as in EBI metagenomics¶
The tools used in the EBI Metagenomics pipeline are also available in ASaiM. We integrate then also a workflow with the same steps as the EBI Metagenomics pipeline (3.0).
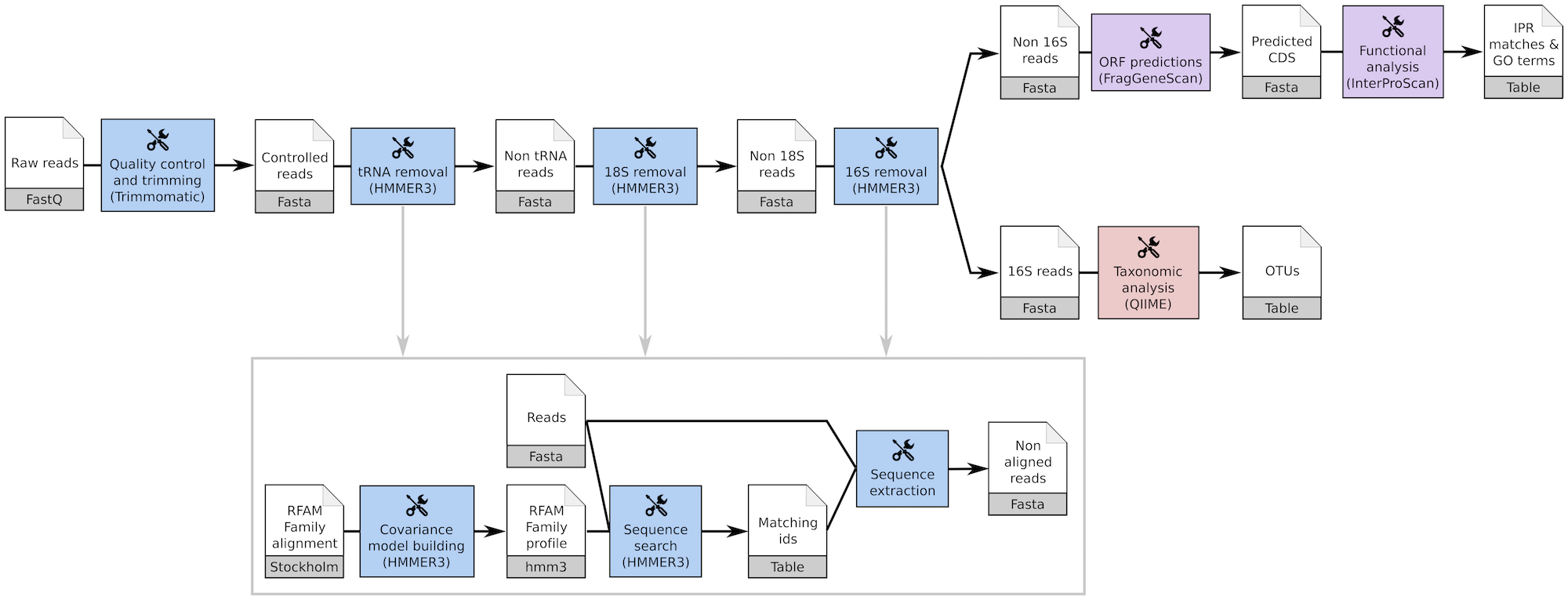
EBI Metagenomics workflow (3.0) in ASaiM
Analyses made in EBI Metagenomics website can be then reproduced locally, without having to wait for availability of EBI Metagenomics or to upload any data on EBI Metagenomics. However the parameters must be defined by the user as we can not find them on EBI Metagenomics documentation.
References
| [1] | Sahar Abubucker, Nicola Segata, Johannes Goll, Alyxandria M. Schubert, Jacques Izard, Brandi L. Cantarel, Beltran Rodriguez-Mueller, Jeremy Zucker, Mathangi Thiagarajan, Bernard Henrissat, Owen White, Scott T. Kelley, Barbara Methé, Patrick D. Schloss, Dirk Gevers, Makedonka Mitreva, and Curtis Huttenhower. Metabolic Reconstruction for Metagenomic Data and Its Application to the Human Microbiome. PLoS Comput Biol, 8(6):e1002358, June 2012. URL: http://dx.doi.org/10.1371/journal.pcbi.1002358, doi:10.1371/journal.pcbi.1002358. |
| [2] | Francesco Asnicar, George Weingart, Timothy L Tickle, Curtis Huttenhower, and Nicola Segata. Compact graphical representation of phylogenetic data and metadata with graphlan. PeerJ, 3:e1029, 2015. |
| [3] | (1, 2, 3) Sherine Awad, Luiz Irber, and C Titus Brown. Evaluating metagenome assembly on a simple defined community with many strain variants. bioRxiv, pages 155358, 2017. |
| [4] | Philip Ewels, Måns Magnusson, Sverker Lundin, and Max Käller. Multiqc: summarize analysis results for multiple tools and samples in a single report. Bioinformatics, 32(19):3047–3048, 2016. |
| [5] | (1, 2) William W Greenwald, Niels Klitgord, Victor Seguritan, Shibu Yooseph, J Craig Venter, Chad Garner, Karen E Nelson, and Weizhong Li. Utilization of defined microbial communities enables effective evaluation of meta-genomic assemblies. BMC genomics, 18(1):296, 2017. |
| [6] | Ben Langmead, Cole Trapnell, Mihai Pop, and Steven L Salzberg. Ultrafast and memory-efficient alignment of short dna sequences to the human genome. Genome biology, 10(3):R25, 2009. |
| [7] | Dinghua Li, Chi-Man Liu, Ruibang Luo, Kunihiko Sadakane, and Tak-Wah Lam. Megahit: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de bruijn graph. Bioinformatics, 31(10):1674–1676, 2015. |
| [8] | Alla Mikheenko, Vladislav Saveliev, and Alexey Gurevich. Metaquast: evaluation of metagenome assemblies. Bioinformatics, 32(7):1088–1090, 2015. |
| [9] | Sergey Nurk, Dmitry Meleshko, Anton Korobeynikov, and Pavel A Pevzner. Metaspades: a new versatile metagenomic assembler. Genome Research, 27(5):824–834, 2017. |
| [10] | (1, 2, 3) Nathan D Olson, Todd J Treangen, Christopher M Hill, Victoria Cepeda-Espinoza, Jay Ghurye, Sergey Koren, and Mihai Pop. Metagenomic assembly through the lens of validation: recent advances in assessing and improving the quality of genomes assembled from metagenomes. Briefings in Bioinformatics, pages bbx098, 2017. |
| [11] | Brian D Ondov, Nicholas H Bergman, and Adam M Phillippy. Interactive metagenomic visualization in a web browser. BMC bioinformatics, 12(1):385, 2011. |
| [12] | Christopher Quince, Alan W Walker, Jared T Simpson, Nicholas J Loman, and Nicola Segata. Shotgun metagenomics, from sampling to analysis. Nature Biotechnology, 35(9):nbt–3935, 2017. |
| [13] | Torbjørn Rognes, Frédéric Mahé, Tomas Flouri, Daniel McDonal, and Pat Schloss. Vsearch: VSEARCH 1.4.0. 2015. URL: https://github.com/torognes/vsearch. |
| [14] | (1, 2) Alexander Sczyrba, Peter Hofmann, Peter Belmann, David Koslicki, Stefan Janssen, Johannes Droege, Ivan Gregor, Stephan Majda, Jessika Fiedler, Eik Dahms, and others. Critical assessment of metagenome interpretation- a benchmark of computational metagenomics software. Biorxiv, pages 099127, 2017. |
| [15] | Duy Tin Truong, Eric A. Franzosa, Timothy L. Tickle, Matthias Scholz, George Weingart, Edoardo Pasolli, Adrian Tett, Curtis Huttenhower, and Nicola Segata. MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat Meth, 12(10):902–903, October 2015. URL: http://www.nature.com.gate1.inist.fr/nmeth/journal/v12/n10/full/nmeth.3589.html, doi:10.1038/nmeth.3589. |
| [16] | (1, 2, 3) Andries Johannes van der Walt, Marc Warwick Van Goethem, Jean-Baptiste Ramond, Thulani Peter Makhalanyane, Oleg Reva, and Don Arthur Cowan. Assembling metagenomes, one community at a time. bioRxiv, pages 120154, 2017. |
| [17] | (1, 2) John Vollmers, Sandra Wiegand, and Anne-Kristin Kaster. Comparing and evaluating metagenome assembly tools from a microbiologist’s perspective-not only size matters! PloS one, 12(1):e0169662, 2017. |